


Unclogging the human data bottleneck
Ideas for improving drug trials




Summary
Trials are the most abundant source of human data, shedding light on how interventions affect us.
But they currently capture much less data than is optimal and lock it up way too tightly.
Collecting 10x more data per trial is possible with ~10% increase in per person cost, enabled through cheap, self-administered at-home tests.
We propose a hypothetical structure to fund collection of this data and make it available for use by other researchers and drug developers.
We then briefly look at an example, the RECOVER trial for understanding long COVID, and outline what a revamped RECOVER could look like under our methodology.
From here, we go more galaxy-brained and explore how this data could be used to build multimodal foundation models for human data, a practical step towards moving beyond simplistic concepts of health and disease.
We also acknowledge that we’ve sketched a loose proposal, but that there are a lot of remaining obstacles to solve in order to make something like this a reality. We discuss cost and privacy as two major ones.
Introduction
In drug discovery, human data is king. It’s a precious, scarce resource that companies pay to acquire and carefully guard, without any guarantee of successful outcome. However, it’s also a public good. Pharma companies don’t fully capture the value accrued from running trials and collecting human data. As a result, trials currently collect fewer types of data and make the data much less accessible than would be optimal for globally accelerating drug development and progress in biology.
Imagine, instead, what it would be like if we had 10 times the amount of available human data, made available in a privacy-preserving way, like a UK Biobank except for all human clinical trial data. Formed in 2006, the UK Biobank (UKBB) collects regular genetic, lifestyle, and health data via the urine, blood and saliva of nearly half a million UK residents. This powerful resource gives researchers pairs of datapoints and health states that help clarify the relationship between particular metrics and health outcomes. Can we scheme up a way of getting more of this high impact data?
Expanded clinical trials present a fantastic opportunity to enhance research data. Trials as they exist today already serve as one of the richest sources of human data. With more than 400,000 active clinical trials globally, each with dozens to thousands of individual participants, there is incredible value to leveraging ongoing trials opportunistically to collect more information from millions of people. More important than data richness is having actual human data. When a first–in-human trial reads out for a new drug category, whole swathes of pharma pay attention, and shift their drug pipeline accordingly. Every trial produces valuable, though selective, data on how interventions translate from animals to humans. But rather than treat trials like the invaluable sources of data they are and capture every last tidbit of information possible, we focus measurement narrowly, letting most of the information they produce slip by unmeasured.
There are many different reasons why trials may not measure more things (hat tip to Alex Telford, who educated us on many of these reasons):
Trial designers understandably tend to be conservative and this just isn’t “how things are done”
It costs extra $
Additional data may even risk becoming a liability
More testing adds to the cost of already-expensive trials
Any measurement that requires provider time adds to total cost
Patients prefer to limit the amount of time and effort required of them as part of trial participation
Learning that a drug works only on a subset of patients positive for a certain measured marker could limit the market of a drug (as it probably should)
Need to educate CROs on how to perform additional assays/tests/measurements
Such data was not as useful before the age of ML methods that can handle high dimensional, heterogeneous inputs
For the moment, rather than focus on any of these reasons in detail, we want to start by sketching out what a future in which we move beyond the current narrow measurement paradigm could look like.
A not-so-modest proposal
As a hypothetical, we’re proposing an industry-wide standardized framework for dataset collection in any given trial, regardless of disease process, endpoint or study objective. This involves creating a set of assays that cover an expansive set of currently-useful metrics (updating whenever necessary), broadly covering metabolic markers, omics, and other diagnostic measurements.
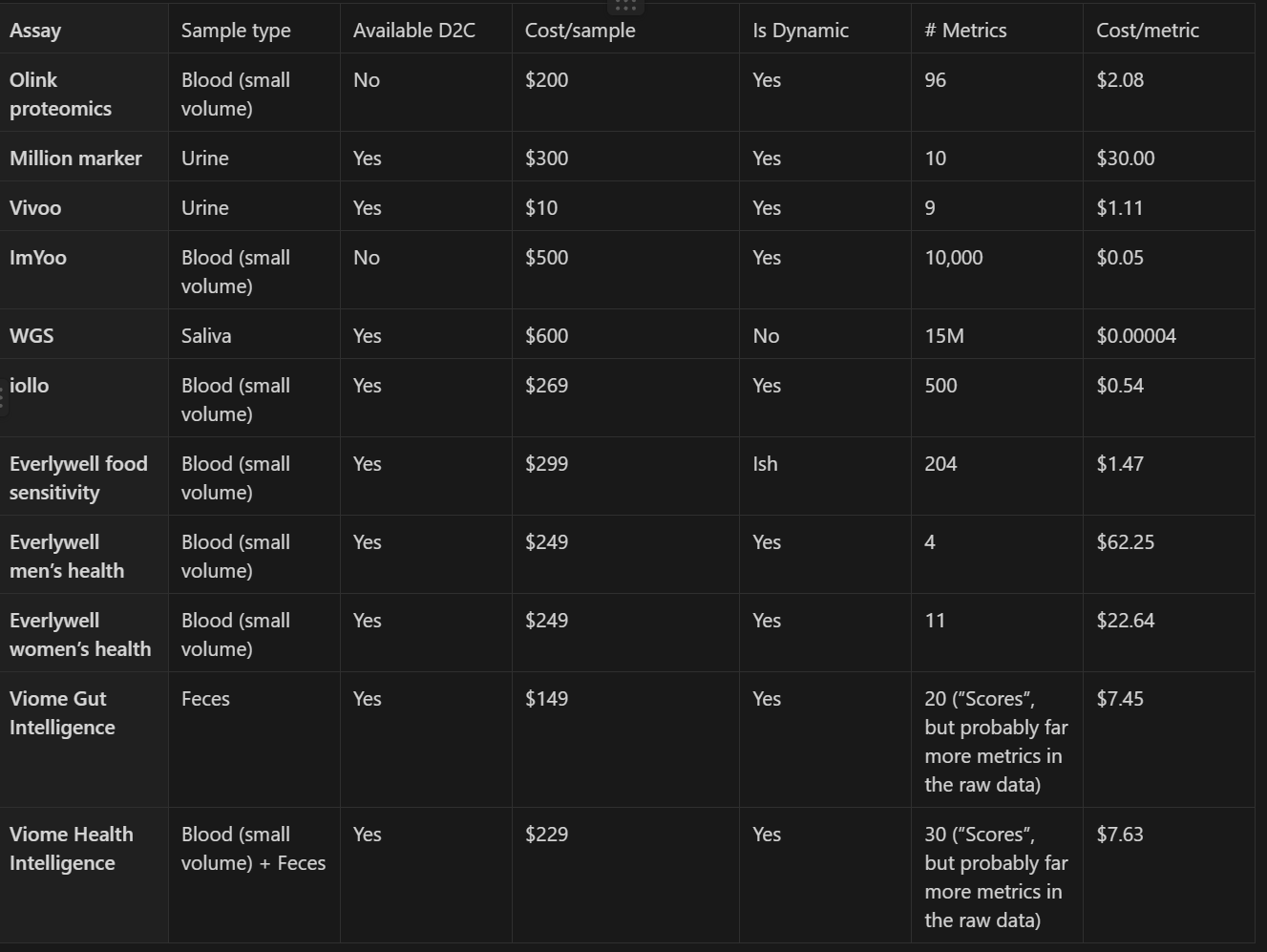
To get a sense of the massive scale of data possible with minimal burden to participants and clinical trial logistics today, the following table proposes biomarkers we could track in every trial and estimates their per sample and per metric costs. This table is only a fraction of all possible non-invasive (i.e. no worse than a blood draw) assays, and is restricted to assays that capture multiple metrics in a single sample, and are able to be performed on samples collected by participants at home without supervision and shipped through conventional mail. We estimate that together these biomarkers would cost ~$3000 per person for one sample of each, so let’s say an upper bound of an additional $6000 per-person per trial for two time points of sample collection. In other words, measuring these biomarkers would add between 10-15% to the average cost of a trial. Also note that these are all current costs, but many of these measurements would get much cheaper if demand increased, allowing for economies of scale and manufacturing learning curves to kick in. All of the proposed tests already allow multiple measurements in one run, and we expect that trend to continue for new tests coming onto the market. Additionally, though our proposal would call for the addition of blood or other sample collection to some types of trials that would otherwise not collect those sample types, for the many drug trials that already do, there would be no additional burden on patients and providers from multiple additional visits.
Collecting a broad spectrum of data in clinical trials, such as immune biomarkers in an Alzheimer’s study, might initially seem extravagant, considering it represents a significant portion of the trial's total cost. This hesitance echoes early doubts in the tech industry about the value of meticulously tracking user interactions within web applications. Critics once deemed it a wasteful consumption of disk space to record every click and scroll, yet this skepticism was shortsighted.
The tech industry’s diligent data collection evolved into a cornerstone for understanding user behavior, leading to innovations and enhanced user experiences. Google, for instance, used extensive data to correlate slight increases in latency with reduced user engagement, spurring efforts to accelerate web speeds. Similarly, Netflix's strategic content creation was underpinned by in-depth analysis of viewer patterns. Translating this to clinical trials, the 'excessive' data gathered today, like immune markers in neurological diseases, may illuminate unforeseen correlations or causal links, potentially revolutionizing our approach to understanding complex disease mechanisms. Just as detailed data collection in tech transformed user experience, so too could expansive clinical data redefine patient treatment, opening new avenues for medical advancement.
Our section on Looking further out takes this a step further and explores if this data could seed the mapping of biological phase space thereby further highlighting the value of collecting multimodal data.
Okay, but who would do this?
Even if you buy our thesis, it only works if a) someone actually collects the data and b) that data is widely accessible to both academic and commercial organizations. We mentioned before some of the myriad reasons current clinical trials may not be taking the shotgun approach of collecting as much data as possible. And right now, limited scope trial data is mostly locked up inside proprietary company databases, which would totally block its use for early stage discovery and other applications that we propose below. Solving this problem could potentially be mitigated by funding this philanthropically and then creating some sort of agreement that ensures the data can be used broadly while preserving privacy, ala UKBB. Similar to the UKBB, this seems like a worthwhile use of philanthropic money, easily worth its weight in the thousands of papers and discoveries enabled by it.
Finding philanthropic funding to pay for the assays themselves doesn't provide relief to all of the concerns of trial designers, such as the complexity, overhead, liability, and data ownership. That's another reason we restricted our list of biomarkers to ones that could be done through self-sampling: if there's too big of an obstacle to adoption, a philanthropic organization could potentially operate as a parallel data collection stream, with the org running the clinical trial only tangentially involved. Participants could, for example, opt-in to participate in this biobank, receive sampling kits at home, and the biobank directly collects the data. It's unlikely that orgs running clinical trials wouldn't later want that data, at which point the biobank could, say, require them to share their outcome data in order to access. There’s a lot more to figure out with such a model, but with the advent of massive ‘omics data possible from self-collected samples, there are likely to be new, creative solutions to incentive alignment between the public, clinical trial participants, and clinical trial designers.
This isn’t the only model that could work and if someone were to really pursue this, they should absolutely explore multiple options. Some of our early readers suggested interesting other ideas, such as:
Follow UKBB’s lead and allow groups to purchase early access, and then release the data widely after an embargo period.
Have governments subsidize this, since, as we mentioned, it’s a public good.
Try to have a for-profit third party collect the data and sell privacy-preserving access.
All of these options come with their own trade-offs, but realistically experimentation is the key here.
An example
COVID’s long term implications remain unresolved. We understand the acute stage of disease – what causes COVID, and how people are affected – but we are still in the dark on what happens after someone’s made a recovery from acute illness but has chronic systems. The RECOVER trial aims to resolve the unanswered questions of long COVID’s impact on various components of human health, most notably the use of paxlovid for the treatment of long COVID and the disease’s influence on various immunological and neurological processes post-infection.
Long COVID has proven extremely difficult to resolve or even make progress on understanding. We understand COVID’s causes well and how they affect people acutely but are still in the dark on what happens when symptoms continue beyond a few weeks. Long COVID provides a good test case for our proposed trial design framework because it’s proven especially tricky to get to the bottom of and is likely a composite of multiple underlying causes.
The RECOVER trial is a large, well-funded trial aimed at improving our understanding of long COVID and how Paxlovid interacts with it. Below, we sketch out what RECOVER 2.0 would look like if we designed it following our framework rather than the current narrow measurement paradigm.
Maintain RCT structure
For every participant, regardless of treatment arm, collect genomic data (WGS), pre-treatment immunological transcriptomics, and the other data in our table collected at least twice (the more data points the merrier)
Post-trial, release the data in a database, ideally that any researcher can access with zero friction
Our strategy is really just an enhancement on what is already being done. The types of questions that were answerable before remain just easily resolved – did the drug work, does the drug have side effects – with additional gains. As an example, for patients that fail to respond to treatment, an expansive set of collected data allows researchers to look to the ‘omics data for answers as to what drives responsiveness, which might otherwise not be possible without standardized assay inclusion, such as cross-referencing with large existing ‘omics + perturbation datasets.
Looking further out
Suppose our post works at persuading people beyond our wildest dreams. Going forward, every clinical trial collects and shares data from all the assays listed in our table and more. Two years go by and we now have a dataset with millions of data points across many different trials rather than just one.
What might the extremely ML-brained way of feeding this back into early discovery efforts look like? Imagine we have a large dataset of trials with data points that look like:
Participant information
Trial and intervention description
Participant assignment
Assay 1 readouts (potentially at multiple time points)
Assay 2 readouts
…
Assay N readouts
Assays could include ‘omics data, CGM measurements, subjective mood ratings, primary trial endpoints, etc. We either initialize a from-scratch model or potentially use a pretrained one and then train it to predict the assay readouts conditional on the participant and trial information across all the trials in the database. For high-dimensional assay like ‘omics, maybe we predict discrete embedding tokens just like they do for images.
Similar to what has been seen in language, proteins, single cell genomics, time series, and other areas, in learning to predict (i.e. compress) the data, our model learns to extrapolate to predicting outcomes when given novel inputs and features that capture underlying biology. It allows us to go beyond concepts of health and disease to map the high dimensional phase space in which each of our biologies live.
More practically, for early drug discovery efforts, we can use this model to predict how novel small molecules or other interventions will affect these readouts. This model doesn’t have to be a perfect predictor to be useful either. As long as the model generates better, predictively valid hypotheses to test in the lab, it would be useful to researchers looking for promising interventions to try.
We could also use this model for target identification. Instead of predicting the effect of a novel compound, here we’d take advantage of the fact that our model’s presumably seen how lots of different interventions impact omics and other readouts. To identify novel targets, we could therefore condition the model on a potential intervention that we know perturbs a target phenotype (but don’t know the mechanism for) and observe what changes in the predicted readouts to identify potential novel targets.
Finally, this data could be used as a key source of target data for transfer learning from animal data (which is much more abundant). This could allow us to bridge the predictive validity gap, potentially enabling much better prediction of trial success and contributing to the war against Eroom’s Law.
This all probably sounds quite fantastical, but the good news is that we don’t have to go straight from nothing to TrialGPT. We could start with narrower prediction problems using subsets of trials and readouts where we expect simpler methods to work, still get value, and then over time build towards this foundation model vision.
The hard stuff
This is a hard thing to get right, and we certainly don’t want to give the impression that system-wide changes that fundamentally change how trials are done can happen easily or that we have a foolproof plan. We are ultimately proposing substantial change to a longstanding system, and there are serious considerations that warrant thought if something like this were to be implemented. To start, adding a standard set of tests covering multiple mediums – mostly blood and urine – increases overhead to already-expensive trials. As we mentioned earlier, the raw cost of the tests alone would add 10-15% to trials which, by itself, presents a large hurdle to overcome, though like most technology we think the per-test costs will go down in time. With those added tests comes increased reliance on participating patients to follow instructions for timing and test collection. Though we think that at-home testing can ameliorate the burden placed on patients, mostly by cutting down on travel time and hospital/clinic-based testing, it’s a big ask to expect stressed and sick patients to follow guidelines strictly. It’s well demonstrated that patients struggle to adhere to required treatment protocols for varying reasons, with developed country adherence hovering around 50%, so it isn’t a stretch to expect that trend to extend to independent data collection regimens.
Moving past logistical hurdles, the other big unanswered question is how to handle data privacy. It isn’t a complete answer, but the UKBB serves as a good example of what proper data governance looks like, though it’s worth mentioning that participants in UKBB data collection are on a strictly opt-in basis. Since we’re proposing adding features to all trials, we’re open to the idea that enhanced collection would happen on an opt-in basis, though that presents challenges with regards to data richness and completeness. Considering all these factors, we should view the additional data as a contribution to public good rather than solely serving commercial interests, which is a vital perspective in assessing the overall value and feasibility of our proposal.
Conclusion
We’ve gone and done something everyone tells you not to do, propose changes to the clinical trials process. The reason everyone tells you not to do this is because major changes to the trials process are risky, require political will, and realistically take many years. So how could this ever possibly be worth it?
For years, people have talked about how our reductionist approach to medicine is hitting diminishing returns. The original pitch for genomic-informed precision medicine spawned out of this perspective over two decades ago, and yet precision, personalized medicine has underperformed its promise.
Recently, multiple people have taken inspiration from AI progress to inform a revitalized perspective on precision medicine and the future of health. We are huge fans of the intersection of these visions, but fear that they’ll never come to pass unless we find ways to acquire and make the right kinds of (human) data available. Although it’s hard, maybe even impossible, opening the clinical trial data faucet is one of the most promising directions for progress in our overarching goal of speeding up biology’s (decades) long development cycle.
Acknowledgements
Thanks to
, Willy and Neal for helpful comments and early feedback. | A guest post by
|
 | A guest post by
|